
Ang lahat ng nilalaman ng iLive ay medikal na nasuri o naka-check ang katotohanan upang masiguro ang mas tumpak na katumpakan hangga't maaari.
Mayroon kaming mahigpit na mga panuntunan sa pag-uukulan at nag-uugnay lamang sa mga kagalang-galang na mga site ng media, mga institusyong pang-akademikong pananaliksik at, hangga't maaari, ang mga pag-aaral ng medikal na pag-aaral. Tandaan na ang mga numero sa panaklong ([1], [2], atbp) ay maaaring i-click na mga link sa mga pag-aaral na ito.
Kung sa tingin mo na ang alinman sa aming nilalaman ay hindi tumpak, hindi napapanahon, o kung hindi pinag-uusapan, mangyaring piliin ito at pindutin ang Ctrl + Enter.
Ang genetic at metabolic na aspeto ng pathogenesis ng osteoarthritis
Medikal na dalubhasa ng artikulo

Huling nasuri: 08.07.2025
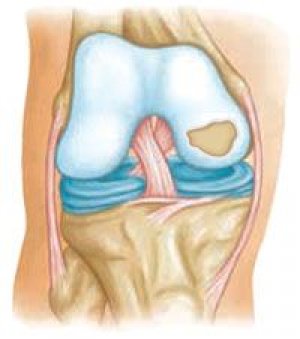 ">
">Ang papel ng mga mekanikal na kadahilanan sa pathogenesis ng osteoarthritis ay hindi maikakaila, ngunit may nakakumbinsi na katibayan na ang ilang mga anyo ng osteoarthritis ay minana ayon sa mga batas ni Mendel. Ang namamana na osteoarthropathies ay maaaring nahahati sa:
- pangunahing pangkalahatang osteoarthritis (PGAO),
- mga arthropathies na nauugnay sa kristal,
- napaaga na osteoarthritis dahil sa namamana na osteochondrodysplasia.
Noong 1803 inilarawan ni W. Heberden ang "medyo siksik na mga node, ang laki ng isang maliit na gisantes" sa dorsal surface ng distal interphalangeal joints ng mga kamay. Ang sintomas na ito, ayon sa may-akda, ay nakikilala ang osteoarthritis mula sa iba pang magkasanib na sakit, kabilang ang gout. Pinalawak ni J. Hayagarth (1805) ang klinikal na paglalarawan ng mga node ni Heberden, na binabanggit ang kanilang madalas na kaugnayan sa arthrosis ng iba pang mga lokalisasyon. Nang maglaon ay inilarawan ni Bouchard ang mga katulad na node sa dorsal surface ng proximal interphalangeal joints ng mga kamay. Gamit ang terminong "Heberden's and Bouchard's nodes", tinukoy ni W. Osier ang "hypertrophic arthritis" at "deforming arthritis" (1909). Noong 1953, natuklasan nina RM Stecher at H. Hersh ang paglaganap ng mga node ni Heberden sa mga miyembro ng pamilya at napagpasyahan na ang mga ito ay minana sa isang autosomal na nangingibabaw na paraan. Ang mga kasunod na pag-aaral kasunod ng pagtuklas nina RM Stecher at H. Hersh ay nagsiwalat ng kaugnayan ng Heberden's at Bouchard's nodes na may degenerative lesions ng iba pang joints. Batay sa data ng klinikal na pagsusuri at pag-type ng HLA, JS Lawrence (1977), JS Lawrence et al. (1983) iminungkahi ang pagkakaroon ng polygenic inheritance sa halip na isang solong depekto ng gene.
Ang phenotypic spectrum ng hereditary osteoarthritis ay malawak na nag-iiba-iba mula sa mga banayad na anyo na nagiging klinikal na nakikita lamang sa huling bahagi ng pagtanda hanggang sa napakatinding anyo na makikita sa pagkabata. Ayon sa kaugalian, ang lahat ng mga form na ito ay inuri bilang pangalawang osteoarthritis. Alam na ngayon na ang ilan sa mga phenotype na ito ay sanhi ng mga mutasyon sa mga gene na naka-encode ng mga macromolecule ng articular cartilage ECM, na nakakagambala sa integridad ng cartilage matrix at ang regulasyon ng paglaganap ng chondrocyte at pagpapahayag ng gene. Ang mga namamana na sakit na ito ay kumakatawan sa isang natatanging subgroup ng osteoarthritis na naiiba sa pangalawang osteoarthritis.
Mga pagkakaiba sa pagitan ng namamana at pangalawang osteoarthritis (ayon kay Williams CJ at Jimenez SA, 1999)
Namamana na osteoarthritis |
Pangalawang osteoarthritis |
|
Etiology |
Mutation ng mga gene na ipinahayag sa articular cartilage |
Iba't ibang namamana at nakuhang sakit |
Pathogenesis |
Pinsala sa mga istruktura o functional na bahagi ng articular cartilage |
Ang mga pangalawang pagpapakita ng sakit, na hindi palaging nakakaapekto lamang sa articular cartilage |
Paggamot |
Maaaring posible ang therapy sa gene upang itama ang depekto ng gene |
Paggamot ng pinagbabatayan na sakit |
Ang Chondrodysplasia/osteochondrodysplasia ay isang pangkat ng mga clinically heterogenous na sakit na nailalarawan ng mga abnormalidad sa paglaki at pag-unlad ng articular cartilage at growth plate. Ang ilang CD/OCD ay humahantong sa maagang pag-unlad ng osteoarthritis, na klinikal na nailalarawan sa pamamagitan ng isang malubhang kurso. Kabilang sa mga ito, ang mga sumusunod na sakit ay maaaring makilala:
- spondyloepiphyseal dysplasia (SED),
- Stickler syndrome,
- dysplasia Knista,
- maramihang epiphyseal dysplasia (MED),
- metaphyseal chondrodysplasia (MCD),
- ilang oto-spondylo-meta-epiphyseal dysplasias (OSMED).
Hereditary dysplasias na nailalarawan sa maagang pagsisimula ng osteoarthritis (ayon kay Williams CJ at Jimenez SA, 1999)
Sakit |
Locus |
Uri ng mana |
Mutated na gene |
Uri ng mutation |
Maagang OA na may late na simula SED (OAR)* |
12q13.1-q13.2 |
IMPYERNO |
COL 2 A, |
Base substitution, insertion, pagtanggal |
Stickler syndrome (STL1) |
12q13.1-q13.2 |
IMPYERNO |
COL2A1 |
Pagpapalit ng base, pagpasok |
Stickler syndrome (STL2) |
6р21.3 |
IMPYERNO |
COLA |
Pagsingit, pagtanggal |
Stickler syndrome |
1p21 |
IMPYERNO |
COLA |
Pagpapalit ng base |
Wagner syndrome |
12q13.1-q13.2 |
IMPYERNO |
COUA, |
Pagpapalit ng base |
OSMED |
6р21.3 |
AR |
COLA |
Pagpapalit ng base |
Marshall syndrome |
1p21 |
IMPYERNO |
COLA |
Ipasok |
Knista dysplasia |
12q13.1-q13.2 |
IMPYERNO |
COLA |
Pagsingit, pagtanggal |
M3fl(EDM1) |
19р13.1 |
IMPYERNO |
COMP |
Pagpapalit ng base |
MED (EDM 2) |
1р32.2-рЗЗ |
IMPYERNO |
COLA |
Ipasok |
MCDS |
6q21-q22.3 |
IMPYERNO |
COLA |
Base substitution, pagtanggal |
MCDJ Jansen |
Зр21.2-р21.3 |
IMPYERNO |
PTHR, |
Pagpapalit ng base |
*Ang mga simbolo ng locus ay ibinibigay sa mga bracket; AD - autosomal na nangingibabaw; AR - autosomal recessive.
Spondyloepiphyseal dysplasia
Kasama sa Spondyloepiphyseal dysplasias (SED) ang isang heterogenous na pangkat ng mga sakit na may autosomal dominant na uri ng mana, na nailalarawan sa abnormal na pag-unlad ng axial skeleton at matinding pagbabago sa epiphyses ng mahabang tubular bones, na kadalasang nagiging sanhi ng dwarfism. Ang SED ay madalas na may malubhang klinikal na kurso, na sinamahan ng pag-ikli ng katawan at, sa isang mas mababang lawak, mga paa.
Sa mga anyo ng EDS na lumilitaw sa mas huling edad, ang phenotype ay kadalasang maliit na nagbabago at maaaring hindi makita sa klinikal hanggang sa pagbibinata, kapag nagkakaroon ng malubhang osteoarthrosis. Ang deformity ng lumbar spine ay maaaring mahayag bilang pagpapaliit ng mga intervertebral disc, platyspondyly, at minor kyphoscoliosis. Ang mga anomalya ng epiphyses sa peripheral joints at maagang degenerative na pagbabago sa mga ito ay napansin din. Ang pinaka-pare-parehong tanda ng peripheral joint damage ay ang pagyupi ng articular surface ng bukung-bukong at mga joint ng tuhod, pati na rin ang pagyupi ng intercondylar groove ng femur. Ang mga anomalya ng ulo at leeg ng femur ay madalas na napansin sa pagbuo ng osteoarthrosis ng hip joint, na nagpapakita sa pagbibinata.
Dahil ang type II collagen ay ang pangunahing bahagi ng hyaline cartilage ECM, iminungkahi na ang gene na naka-encode nito, COL1A, ay ang sanhi ng EDS. Ang unang paglalarawan ng isang genetic na link sa pagitan ng phenotype ng maagang osteoarthritis na nauugnay sa late-onset na EDS at ang procollagen type II gene, COL 2 A, ay nagsimula noong 1989 at 1990. Ang unang ulat ng isang COL 2 A mutation sa mga kamag-anak na may maagang osteoarthritis na nauugnay sa late-onset na EDS ay kasama ang Arg519>Cys base substitution. Sa ngayon, apat pang pamilya na may katulad na mutasyon ang natukoy. Sa mga miyembro ng isa pang pamilya na may maagang OA at banayad na EDS, natagpuan ang Arg75>Cys base substitution, bagama't ang EDS phenotype sa mga miyembro ng pamilyang ito ay hindi katulad ng phenotype ng pamilya na may arginine sa cysteine substitution sa posisyon na 519. Iba pang mga mutasyon na COL 2 A-Gly976>Ser> sa mga miyembro ng pamilyang EDS ay kasama rin sa mga miyembro ng EDS na si Gly493. J. Spranger et al. (1994) ginamit ang terminong "type 11 collagenopathy" upang ilarawan ang mga namamana na sakit ng cartilage tissue na may pangunahing mutation sa procollagen type II gene COL1A.
Klasikong anyo ng Stickler syndrome
Una itong inilarawan noong 1965 ni GB Stickler at mga kasamahan, na tinawag itong hereditary arthro-ophthalmopathy. Ang sindrom na inilarawan ng GB Stickler ay nailalarawan sa pamamagitan ng visual impairment at malubhang degenerative joint disease, na kadalasang nabubuo sa ikatlo o ikaapat na dekada ng buhay. Ito ay isang autosomal dominant disorder na may saklaw na humigit-kumulang 1 sa 10,000 live births. Kasama sa clinical presentation ang myopia, progressive deafness, cleft palate, hypoplasia ng mandible (Pierre-Robin anomaly), at hypoplasia ng epiphyses. Sa panahon ng neonatal, ang mga radiograph ng mga pasyente na may Stickler syndrome ay nagpapakita ng mga pinalaki na epiphyses, pangunahin ang proximal femur at distal tibia. Sa panahon ng paglaki, ang epiphyseal dysplasia ay bubuo, na ipinakita sa pamamagitan ng hindi regular na ossification ng epiphyses at kasunod na mga degenerative na pagbabago.
Dahil ang COL 2 A ay ipinahayag sa articular cartilage at ang vitreous body ng eyeball, ang paglitaw ng Stickler syndrome ay nauugnay sa patolohiya ng gene na ito. Gayunpaman, ang pagsusuri sa ilang pamilyang may Stickler syndrome ay nagpakita na hindi lahat ng pamilya ay may sakit na nauugnay sa COL 2 A. Ang uri ng sakit na ito ay tinatawag na type I Stickler syndrome (locus symbol STL1).
Ang spectrum ng mga klinikal na pagpapakita ng Stickler syndrome ay malawak na nag-iiba, at ilang mga phenotypes ang natukoy hanggang sa kasalukuyan. Kabilang sa mga ito ay Wagner syndrome, na kung saan ay nailalarawan sa pamamagitan ng isang pamamayani ng pinsala sa eyeball; Ang OA sa Wagner syndrome ay halos hindi nagkakaroon, bagama't may natukoy na mutation ng COL 2 A gene (base substitution Gly67>Asp) sa mga pasyente. Ito ay nananatiling hindi maliwanag kung bakit ang gayong COL mutation ay nakompromiso lamang ang paggana ng vitreous body at hindi nakakaapekto sa hyaline cartilage.
Ang isa pang anyo ng Stickler syndrome ay ang tinatawag na Dutch variant; ito ay nailalarawan sa lahat ng mga klasikong pagpapakita ng sindrom maliban sa kapansanan sa paningin. HG Brunner et al. (1994) ay nagpakita na ang Dutch phenotype ng Stickler syndrome ay nauugnay sa isang mutation sa COL,,A 2 gene: ang nangingibabaw na mutation ay isang 54-base na pagtanggal ng pares na sinusundan ng isang exon deletion. M. Sirko-Osadsa et al. (1998) ay nag-ulat ng isa pang pamilya, na hindi nauugnay sa isang inilarawan ng mga nakaraang may-akda, na may katulad na phenotype at isang mutation sa COL,,A 2 gene (27-base na pagtanggal ng pares), na nagpapatunay sa data ng HG Brunner et al. (1994). Ang variant na ito ay tinatawag na type II Stickler syndrome (locus symbol STL1).
Kamakailan, natukoy ang ikatlong locus ng Stickler syndrome sa mga miyembro ng isang pamilya na may vitreous at retinal pathology na phenotypically makabuluhang naiiba mula sa mga pagbabagong naobserbahan sa "classic" na variant ng syndrome. Isang mutation sa COL2A| gene (base substitution Gly97>Val) ay natagpuan sa mga miyembro ng pamilyang ito. Siyempre, ang mga bagong paglalarawan ng mga kaso ng pheno- at genotype na ito ng Stickler syndrome ay kinakailangan upang kumpirmahin ang mga natuklasan ng AJ Richards et al.
Ang nosological na koneksyon sa pagitan ng Marshall syndrome at ang klasikong bersyon ng Stickler syndrome ay tinalakay nang mahabang panahon. Ngayon ang Marshall syndrome ay inuri bilang isang hiwalay na phenotype higit sa lahat dahil sa mas malinaw na pagpapapangit ng facial skeleton, bagaman ang pinsala sa peripheral joints ay katulad ng sa type I Stickler syndrome. Sa Marshall syndrome, ang osteoarthritis ng mga kasukasuan ng tuhod at lumbosacral spine ay nagsisimula pagkatapos ng 30 taon. Ang sanhi ng sindrom ay isang mutation sa type IX collagen gene COL n A1.
 [ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ]
[ 1 ], [ 2 ], [ 3 ], [ 4 ], [ 5 ], [ 6 ]
OSMED
Ang phenotype na ito ay inilarawan sa isang pamilyang Dutch kung saan ang mga degenerative na pagbabago sa mga joints na kahawig ng osteoarthrosis ay lumitaw sa pagbibinata at apektado pangunahin ang hip, tuhod, siko at balikat; Ang mga kakaibang tampok ng mukha, nadagdagan ang lumbar lordosis, pinalaki na mga interphalangeal joints, at pagkawala ng pandinig ay natagpuan din, ngunit walang nakitang anomalya sa paningin (Vikkula M. et al., 1995). Natuklasan ng mga mananaliksik ang isang mutation sa gene na naka-encode sa isang 2 -chain ng type II collagen COL,,A 2.
Knista dysplasia
Nailalarawan sa pamamagitan ng pagpapaikli ng trunk at limbs, pagyupi ng mukha at tulay ng ilong, exophthalmos, at malubhang joint abnormalities. Sa mga pasyente na may Kniest syndrome, ang mga joints, kadalasang malaki mula sa kapanganakan, ay patuloy na lumalaki sa pagkabata at maagang pagbibinata. Madalas din silang may myopia, pagkawala ng pandinig, cleft palate, at clubfoot; karamihan sa mga pasyente ay nagkakaroon ng malubhang degenerative na pagbabago nang maaga, lalo na binibigkas sa mga kasukasuan ng tuhod at balakang. Ang spinal radiographs ay nagpapakita ng pagyupi at makabuluhang pagpahaba ng mga vertebral na katawan at platyspondyly. Ang mahabang tubular bones ay deformed tulad ng isang dumbbell, at ang ossification ng epiphyses ay mabagal. Sa mga joints ng mga kamay, ang mga epiphyses ay pipi at ang magkasanib na mga puwang ay makitid. Ang articular cartilage ay malambot, ang pagkalastiko nito ay nabawasan; histologically, ang malalaking cyst ay matatagpuan dito (ang sintomas ng "Swiss cheese"). Ang Kniest syndrome ay sanhi ng mutation sa procollagen type II gene COb2A1.
[ 7 ], [ 8 ], [ 9 ], [ 10 ], [ 11 ], [ 12 ], [ 13 ], [ 14 ]
Maramihang epiphyseal dysplasia (MED)
Isang heterogenous na pangkat ng mga sakit na nailalarawan sa abnormal na pag-unlad ng mga plate ng paglago ng mahabang tubular bones, pati na rin ang maagang (nagpapakita sa pagkabata) malubhang osteoarthrosis na nakakaapekto sa parehong axial at peripheral joints (madalas na tuhod, balakang, balikat at kamay). Sa klinika, ang MED ay nagpapakita ng sarili bilang sakit at paninigas sa mga kasukasuan, mga pagbabago sa lakad. Ang mga pasyente na may MED ay mayroon ding kaunting pagbabago sa spinal column (iba't ibang antas ng pagyupi ng mga vertebral na katawan), kung minsan ang gulugod ay buo. Ang maikling tangkad ng mga pasyente ay katangian din, bagaman ang dwarfism ay bihirang bubuo. Hindi apektado ang visual organ. Kasama sa MED ang ilang variant, halimbawa, ang Fairbanks at Ribbing phenotype.
Ang mga MED ay minana sa autosomal dominant na paraan na may iba't ibang antas ng penetrance. Dahil ang tanda ng MED ay isang anomalya ng epiphyseal growth plate, iminungkahi na ang mga dysplasia na ito ay sanhi ng isang depekto sa mga gene na naka-encode ng mga macromolecule ng growth plate cartilage. Ito ay lumabas na hindi bababa sa tatlong loci ang nauugnay sa MED phenotype. Pag-aaral ni EJ Weaver et al. (1993), JT Hecht et al. (1992) ay hindi kasama ang mga gene ng mga uri ng collagen II at VI, ang pangunahing protina ng mga proteoglycan, at ang nag-uugnay na protina ng kartilago mula sa listahan ng mga "salarin" ng mga MED. JT Hecht et al. (1993), R. Oehelmann et al. (1994) ay natagpuan ang isang link sa pagitan ng MED, pati na rin ang clinically related pseudoachondroplasia syndrome, at ang pericentromeric region ng chromosome 19. Natukoy ng mga kasunod na pag-aaral ang isang mutation sa gene encoding cartilage oligomeric matrix protein (OMMP) sa tatlong pasyente na may MED (locus symbol EDM1). Dahil ang lahat ng tatlong mutasyon ay naganap sa rehiyon ng gene na naka-encode ng calcium-binding domain ng OMMP, malamang na ang calcium-binding function ng protina na ito ay mahalaga para sa normal na pag-unlad ng growth plate cartilage.
MD Briggs et al. (1994) ay nag-ulat ng isang pamilyang Dutch na may MED phenotype na nauugnay sa isang rehiyon ng chromosome 1 na naglalaman ng isa sa uri ng IX collagen genes, COL1A1 (simbolo ng EDM 2 locus). Kapansin-pansin, ang mutation na natagpuan ay ang unang katibayan ng isang papel para sa type IX collagen, na naisalokal sa ibabaw ng collagen II fibrils, sa pagpapanatili ng integridad ng hyaline cartilage. M. Deere et al. (1995) ay nagpakita na ang Fairbanks phenotype ay hindi genetically na nauugnay sa alinman sa EDM o EDM2 locus, na nagpapatunay sa heterogeneity ng MED.
Metaphyseal chondrodysplasia (MCD)
Ang isang heterogenous (higit sa 150 mga uri ay inilarawan) na pangkat ng mga namamana na sakit ng hyaline cartilage, na clinically manifest bilang maagang osteoarthrosis. Ang mga MHD ay nailalarawan sa pamamagitan ng mga pagbabago sa metaphyses ng buto. Sa klinika, ang mga ito ay nagpapakita bilang maikling tangkad, pinaikling mga paa, nakayukong mga sandalyas, at isang "duck" na lakad. Ang mga pasyente na may MHD ay nagpapakita rin ng mga palatandaan ng pinsala sa ibang mga sistema (halimbawa, ang immune at digestive system). Ang disorganization ng growth plate cartilage ay sinusunod, na histologically manifests mismo bilang mga kumpol ng proliferated at hypertrophied chondrocytes na napapalibutan ng thickened septa at disorganized matrix, pati na rin ang pagtagos ng non-calcified cartilage sa subchondral bone.
Ang mga Jansen, Schmid at McKusick syndrome ay ang pinaka-pinag-aralan na mga MHD. Ang mga ito ay katulad sa mga tampok ng skeletal anomalya, ngunit naiiba sa kalubhaan (Jansen syndrome-McKusick syndrome-Schmid syndrome). Ang pinakakaraniwan ay ang Schmid syndrome (simbolo ng MCDS locus), na minana sa isang autosomal dominant na paraan. Sa radiologically, ang sindrom ay ipinakita sa pamamagitan ng coxa vara, pagpapaikli at kurbada ng mga tubular na buto, hugis-tasa na pagpapapangit ng mga metaphyses (mas binibigkas sa proximal kaysa sa distal na bahagi ng femur). Ang pinaka-binibigkas na mga pagbabago ay sinusunod sa mga plate ng paglago ng mahabang tubular bones.
Hindi bababa sa 17 iba't ibang uri ng collagen X gene mutations ang inilarawan sa mga pasyenteng may Schmid syndrome. Ang Collagen X ay ipinahayag sa hypertrophied chondrocytes ng growth plates at maaaring kasangkot sa mga proseso ng ossification. Kaya, ang isang mutation sa collagen X gene COb2A1 ay ang pinaka-malamang na sanhi ng Schmid syndrome.
Ang mga batang may Jansen syndrome ay may hypercalcemia, mataas na antas ng urinary phosphate, at pagbaba ng parathyroid hormone (PTH) at mga antas ng peptide na nauugnay sa PT. Ang anomalya ng huli ay malamang na responsable para sa pagbuo ng Jansen syndrome. Noong 1994, inilathala ng AS Karaplis at mga kapwa may-akda ang mga resulta ng isang orihinal na pag-aaral. Pagkatapos ng pagkagambala ng gene na naka-encode ng PT-related peptide sa mouse embryonic stem cells, ang mga daga na may kakulangan sa allele na ito ay namatay kaagad pagkatapos ng kapanganakan. Napag-alaman na mayroon silang anomalya sa subchondral bone development, may kapansanan sa paglaki ng cartilage, at pagbaba ng chondrocyte proliferation. Noong 1995, iniulat ni E. Schipani at ng mga co-authors ang isang heterozygous mutation sa PTH receptor gene sa isang pasyente na may Jansen syndrome. Ang mutation ay binubuo ng isang Gys223>Arg base substitution, na humantong sa cAMP accumulation; Nangangahulugan ito na ang amino acid histidine sa posisyon 223 ay gumaganap ng isang mahalagang papel sa paghahatid ng signal. Nang maglaon, E. Schipani et al. (1996) ay nag-ulat ng tatlong iba pang mga pasyente na may Jansen syndrome, dalawa sa kanila ay may katulad na mutation, at ang pangatlo ay nagkaroon ng TrА10>Ро substitution.
Pangunahing pangkalahatang osteoarthritis
Ang pinakakaraniwang namamana na anyo ng osteoarthritis ay ang primary generalized osteoarthritis (PGOA), na unang inilarawan bilang isang hiwalay na nosology nina JH Kellgren at R. Moore noong 1952. Sa clinically, ang primary generalized osteoarthritis ay nailalarawan sa pamamagitan ng paglitaw ng Bouchard's at Heberden's nodes, polyarticular lesions. Ang pangunahing pangkalahatang osteoarthritis ay nailalarawan sa pamamagitan ng maagang pagsisimula ng pagpapakita ng osteoarthritis at ang mabilis na pag-unlad nito. Sa radiologically, ang primary generalized osteoarthritis ay hindi naiiba sa non-hereditary osteoarthritis. Sa kabila ng katotohanan na ang isyu ng etiopathogenesis ng pangunahing pangkalahatang osteoarthritis ay pinagtatalunan pa rin, ang mga pag-aaral ay nagpapakita ng mahalagang papel ng namamana na predisposisyon sa paglitaw at pag-unlad ng pangunahing pangkalahatang osteoarthritis.
Kaya, JH Kellgren et al. (1963) natagpuan ang mga node ng Boucharay-Heberden sa 36% ng mga lalaking kamag-anak at 49% ng mga babaeng kamag-anak, habang sa pangkalahatang populasyon ang mga bilang na ito ay 17 at 26%, ayon sa pagkakabanggit. Sa mga indibidwal na may pangunahing pangkalahatang osteoarthritis, ang HLA A1B8 haplotype at ang MZ isoform ng a1-antitrypsin ay mas madalas na natutukoy. Sa isang klasikong pag-aaral na kinasasangkutan ng kambal, si TD Spector et al. (1996) ay nagsagawa ng radiography ng mga kasukasuan ng tuhod at mga kasukasuan ng kamay sa 130 monozygotic at 120 na kambal na pangkapatid na babae para sa mga pagbabagong katangian ng osteoarthritis. Ito ay naka-out na ang concordance ng radiographic na mga palatandaan ng osteoarthritis ng lahat ng mga localization ay 2 beses na mas mataas sa monozygotic twins kumpara sa fraternal twins, at ang kontribusyon ng genetic factor ay mula 40 hanggang 70%. Isang pag-aaral ng nodular osteoarthritis ni GD Wright et al. (1997) ay nagpakita ng maagang pagsisimula ng sakit, mataas na kalubhaan, at isang negatibong ugnayan sa pagitan ng edad ng pagsisimula ng sakit sa mga pasyente at ang edad ng paglilihi ng kanilang mga magulang.
Kabilang sa mga arthropathies na nauugnay sa kristal, ang pagtitiwalag ng mga kristal ng uric acid at mga kristal na naglalaman ng calcium sa magkasanib na lukab ay may predisposisyon sa pamilya.
Hereditary crystal-associated arthropathies (ayon kina Williams CJ at Jimenez SA, 1999)
Sakit |
Locus |
Uri ng mana |
Mutated na gene |
Uri ng mutation |
Gout (HPRT)* |
Xq27 |
X-linked |
HPRT1 |
Base substitution, pagtanggal |
Gout (PRPS) |
Xq22-q24 |
X-linked |
PRPS1 |
Pagpapalit ng base |
Pangunahing pyrophosphate arthropathy (CCAL1) |
5р15.1-р15.2 |
IMPYERNO |
? |
? |
Early-onset pyrophosphate arthropathy na nauugnay sa 0A (CCAL2) |
8q |
IMPYERNO |
? |
? |
*Ang mga simbolo ng locus ay ibinibigay sa mga bracket; AD – autosomal dominant.
Noong 1958, ipinakita ni D. Zintann S. Sitaj ang mga klinikal na paglalarawan ng isang patolohiya na tinatawag nilang "chondrocalcinosis" sa 27 mga pasyente. Karamihan sa mga pasyente ay kabilang sa limang pamilya, na nagpapahiwatig ng isang namamana na bahagi sa etiopathogenesis ng sakit. Nang maglaon, iniulat nina D. McCarty at JL Hollander (1961) ang dalawang pasyente na pinaghihinalaang may gout na may deposition ng mga nonurate crystals sa joint cavity. Ang pagsusuri sa X-ray ay nagsiwalat ng abnormal na calcification ng hyaline cartilage ng maraming joints.
Sa radiographically, ang calcium pyrophosphate dihydrate crystal deposition disease, o pyrophosphate arthropathy, ay kahawig ng sporadic OA, ngunit mas madalas itong nakakaapekto sa mga joints na hindi tipikal para sa mga karaniwang anyo ng osteoarthrosis (hal., metacarpophalangeal, scaphoradial, patellofemoral knee joints). Sa pyrophosphate arthropathy, ang mga subchondral bone cyst ay mas madalas na nabuo. Bagaman sa karamihan ng mga kaso, ang chondrocalcinosis ay nangyayari bago ang pagpapakita ng pangalawang osteoarthrosis, sa ilang mga indibidwal ang sakit ay maaaring magsimula bilang idiopathic osteoarthrosis, na sinamahan ng metabolic disorder (hemochromatosis, hyperparathyroidism, hypomagnesemia, atbp.).
Malamang, ang mga pagbabago sa istruktura sa ECM ng articular cartilage ay nag-udyok sa pagtitiwalag ng calcium pyrophosphate dihydrate crystals. Natagpuan ni AO Bjelle (1972, 1981) ang pagbaba sa nilalaman ng collagen at pagkapira-piraso ng mga hibla ng collagen sa gitnang zone ng matrix ng articular cartilage ng mga miyembro ng pamilyang Swedish na may pyrophosphate arthropathy. Dahil ang mga lugar na ito ay hindi naglalaman ng mga kristal, iminungkahi ng mga may-akda na ang inilarawan na matrix anomalya ay maaaring mag-predispose sa kanilang pagtitiwalag at ang pagbuo ng mga degenerative na pagbabago sa mga joints. Batay sa isang pag-aaral ng mga sporadic na kaso ng pyrophosphate arthropathy, K. Ishikawa et al. (1989), I. Masuda et al. (1991) ay napagpasyahan na ang chondrocalcinosis ay sanhi ng isang mutation sa mga gene na naka-encode ng mga protina ng ECM. CJWilliams et al. (1993), AJ Reginato et al. (1994) natagpuan ang isang heterozygous mutation COL 2 A, (base substitution Argl5>Cys) sa mga miyembro ng isang malaking pamilya na may clinical phenotype ng malubhang maagang osteoarthritis na may ankylosis, late development ng spondyloepiphyseal dysplasia at chondrocalcinosis ng hyaline at fibrocartilage. Gayunpaman, lumabas na sa mga miyembro ng pamilyang ito ang chondrocalcinosis ay pangalawa sa OA.
Iminungkahi din na ang mga inorganikong sangkap ng ECM ay nag-aambag sa pagbuo ng kristal. Halimbawa, ang hypomagnesemia ay nagdudulot ng chondrocalcinosis sa pamamagitan ng pagpigil sa enzyme pyrophosphatase, na binabawasan naman ang pagkalusaw ng kristal. Ang mga mataas na antas ng inorganic phosphate ay natagpuan sa synovial fluid ng mga pasyente na may pyrophosphate arthropathy. Ito at iba pang mga obserbasyon ay nagmungkahi na ang mga pasyente na may pyrophosphate arthropathy ay may lokal na karamdaman ng pyrophosphate metabolism. Ang enzyme na nucleoside triphosphate pyrophosphohydrolase ay inilarawan, na maaaring kasangkot sa pagbuo ng mga kristal na pyrophosphate sa lugar ng kanilang pagtitiwalag sa ECM. Ang mataas na antas ng enzyme na ito ay natagpuan sa mga sporadic na kaso ng pyrophosphate arthropathy, ngunit ang abnormalidad na ito ay hindi naobserbahan sa mga familial na anyo ng sakit (Ryan LM et al., 1986). Gayunpaman, kapag ang pag-culture ng mga fibroblast at lymphoblast mula sa mga pasyente na may familial pyrophosphate arthropathy, ang isang pagtaas sa nilalaman ng mga inorganic phosphate ay napansin, na nagpapatunay din sa pagpapalagay tungkol sa papel ng mga kaguluhan sa lokal na metabolismo ng pyrophosphate sa pathogenesis ng sakit.
Sa mga nagdaang taon, ang mga pagtatangka ay ginawa upang makilala ang mga gene na "nagkasala" ng paglitaw ng mga familial na kaso ng pyrophosphate arthropathy. Kaya, ang pagsusuri ng genetic na materyal na nakuha mula sa mga miyembro ng isang malaking pamilya na may pyrophosphate arthropathy (Maine, USA), kung saan ang chondrocalcinosis ay binuo ng pangalawa sa malubha, mabilis na pag-unlad, non-dysplastic osteoarthrosis, hindi kasama ang isang koneksyon sa pagitan ng sakit at COL 2 locus. Gayunpaman, natagpuan ng mga may-akda ng pag-aaral na ito ang isang koneksyon sa pagitan ng pinag-aralan na phenotype ng pyrophosphate arthropathy at isang locus na matatagpuan sa mahabang braso ng chromosome 8 (ang simbolo ng CCAL locus). AG Hughes et al. (1995) ay natagpuan ang isang koneksyon sa pagitan ng phenotype ng pangunahing chondrocalcinosis sa isang pamilya mula sa UK at ang CCAL1 locus, na naisalokal sa maikling braso ng chromosome 5 sa 5p15 na rehiyon. Ayon kay CJ Williams et al. (1996), ang CCAL1 locus sa mga miyembro ng isang pamilyang Argentine na may pyrophosphate arthropathy ay medyo mas malapit kaysa sa nakaraang kaso, sa 5p15.1 na rehiyon. Ang isang katulad na genotype ay natagpuan sa mga miyembro ng isang pamilya mula sa France.
Kaya, ang data mula sa inilarawan na mga pag-aaral ay nagpapahiwatig na ang familial form ng pyrophosphate arthropathy ay isang clinically at genetically heterogenous na sakit, na maaaring sanhi ng mga mutasyon sa hindi bababa sa tatlong magkakaibang mga gene.